
VERZENIOS 150 mg, comprimé pelliculé, boîte de 14
Retiré du marché le : 03/01/2022
Dernière révision : 01/04/2022
Taux de TVA : 10%
Laboratoire exploitant : LILLY FRANCE
Source :

Cancer du sein précoce
Verzenios en association avec une hormonothérapie est indiqué chez les patients adultes en traitement adjuvant du cancer du sein précoce avec récepteurs hormonaux (RH) positifs, récepteurs du facteur de croissance épidermique humain 2 (human epidermal growth factor receptor 2 [HER2]) négatifs, avec atteinte ganglionnaire et haut risque de rechute (voir rubrique Propriétés pharmacodynamiques).
Chez les femmes en pré/périménopause, le traitement par un inhibiteur de l'aromatase comme hormonothérapie doit être associé à un agoniste de l'hormone de libération de la lutéinostimuline (luteinizing hormone-releasing hormone, LHRH).
Cancer du sein avancé ou métastatique
Verzenios est indiqué chez les femmes dans le traitement du cancer du sein localement avancé ou métastatique, avec récepteurs hormonaux (RH) positifs, et récepteurs du facteur de croissance épidermique humain 2 (HER2) négatifs en association avec un inhibiteur de l'aromatase ou avec le fulvestrant comme hormonothérapie en première intention, ou chez les femmes ayant été traitées antérieurement par hormonothérapie.
Chez les femmes en pré/périménopause, l'hormonothérapie doit être associée à un agoniste de la LHRH.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Neutropénie
Des cas de neutropénie ont été rapportés chez des patients traités par abémaciclib. Une modification de la dose est recommandée chez les patients qui développent une neutropénie de grade 3 ou 4 (voir rubrique Posologie et mode d'administration). Des événements à issue fatale de sepsis neutropénique ont eu lieu chez moins de 1 % des patientes atteintes d'un cancer du sein métastatique. Il doit être demandé aux patients de rapporter tout épisode de fièvre à leur médecin, leur pharmacien ou leur infirmier/ère.
Infections/infestations
Des infections ont été rapportées chez des patients traités par abémaciclib en association à une hormonothérapie à un taux plus élevé que chez ceux traités par hormonothérapie. Une infection du poumon a été rapportée chez des patients traités par abémaciclib sans neutropénie concomitante. Des événements à issue fatale ont eu lieu chez moins de 1 % des patientes atteintes d'un cancer du sein métastatique. Il faut surveiller les signes et symptômes d'infection chez les patients et les traiter de façon médicalement appropriée.
Thrombo-embolies veineuses
Des événements thromboemboliques veineux ont été rapportés chez des patients traités par l'association d'abémaciclib avec une hormonothérapie. Il faut surveiller les patients afin de détecter d'éventuels signes et symptômes de thromboses veineuses profondes et d'embolies pulmonaires et traiter de façon médicalement appropriée. En fonction du grade de l'événement thrombo-embolique veineux, il peut être nécessaire de modifier la dose d'abémaciclib (voir rubrique Posologie et mode d'administration).
Élévations des transaminases
Des élévations de l'ALAT et de l'ASAT ont été rapportées chez des patients traités par abémaciclib. En fonction du niveau d'élévation de l'ALAT ou de l'ASAT, il peut être nécessaire de modifier la dose d'abémaciclib (voir rubrique Posologie et mode d'administration).
Diarrhée
La diarrhée est l'effet indésirable le plus fréquent. Dans toutes les études cliniques, le délai médian d'apparition de la première diarrhée était d'environ 6 à 8 jours, et la durée médiane de la diarrhée était de 7 à 12 jours (grade 2) et de 5 à 8 jours (grade 3). La diarrhée peut être associée à une déshydratation. Les patients doivent commencer un traitement avec des agents antidiarrhéiques tels que le lopéramide au premier signe de selles molles, augmenter leur prise de liquides par voie orale et informer leur médecin, leur pharmacien ou leur infirmier/ère. Une modification de la dose est recommandée chez les patients chez lesquels survient une diarrhée ≥ grade 2 (voir rubrique Posologie et mode d'administration).
Pneumopathie interstitielle diffuse (PID)/Pneumonie
Des cas de PID/pneumonie ont été rapportés chez des patients traités par abémaciclib. Surveiller les symptômes pulmonaires évocateurs d'une PID/pneumonie chez les patients et les traiter de façon médicalement appropriée. En fonction du grade de la PID/pneumonie, une modification de la dose d'abémaciclib peut être nécessaire (voir rubrique Posologie et mode d'administration). Arrêter définitivement l'abémaciclib chez les patients présentant une PID/pneumonie de grade 3 ou 4.
Administration concomitante d'inducteurs du CYP3A4
L'administration concomitante d'inducteurs du CYP3A4 doit être évitée en raison du risque de diminution de l'efficacité de l'abémaciclib (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Crise viscérale
Il n'y a pas de données sur l'efficacité et la tolérance d'abémaciclib chez les patients présentant une crise viscérale.
Lactose
Les patients présentant des maladies héréditaires rares d'intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose ou du galactose ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables les plus fréquents sont : diarrhée, infections, neutropénie, leucopénie, anémie, fatigue, nausées, vomissements, alopécie et baisse d'appétit.
Parmi les effets indésirables les plus fréquents, ceux de grade ≥ 3 étaient inférieurs à 5 % à l'exception de la neutropénie, de la leucopénie, et de la diarrhée.
Liste tabulée des effets indésirables
Dans le tableau ci-après, les effets indésirables sont répertoriés selon la classification MedDRA par classes de systèmes d'organes et par fréquence. Les catégories de fréquence sont les suivantes : Très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque catégorie de fréquence, les effets indésirables sont présentés par ordre décroissant de sévérité.
Tableau 8. Effets indésirables rapportés dans les études de phase III de l'abémaciclib en association avec une hormonothérapiea (N = 3 559)
| Classes de Systèmes d'Organes | Très fréquent | Fréquent | Peu fréquent |
| Infections et infestations | Infections b | | |
| Affections hématologiques et du système lymphatique | Neutropénie Leucopénie Anémie Thrombocytopénie Lymphopénie h | | Neutropénie febrile e |
| Troubles du métabolisme et de la nutrition | Baisse d'appétit | | |
| Affections du système nerveux | Céphalée f Dysgueusie g Sensation vertigineuse g | | |
| Affections oculaires | | Augmentation de la sécrétion lacrymale | |
| Affections vasculaires | | Thrombo-embolies veineuses c | |
| Affections respiratoires, thoraciques et médiastinales | | Pneumopathie interstitielle diffuse (PID)/pneumonie d | |
| Affections gastro-intestinales | Diarrhée Vomissements Nausées Stomatites f | Dyspepsie f | |
| Affections de la peau et du tissu sous-cutané | Alopécie g Prurit g Rash g | Troubles des ongles f Sécheresse cutanée e | |
| Affections musculo- squelettiques et systémiques | | Faiblesse musculaire e | |
| Troubles généraux et anomalies au site d'administration | Pyrexie e Fatigue | | |
| Investigations | Élévation de l'alanine aminotransférase g Élévation de l'aspartate aminotransférase g | | |
a Abémaciclib en association avec l'anastrozole, le létrozole, l'exémestane, le tamoxifène ou le fulvestrant.
b Les infections comprennent tous les termes préférentiels (PT) rapportés faisant partie de la Classe de Systèmes d'Organes « Infections et infestations ».
c Les événements thromboemboliques veineux comprennent la thrombose veineuse profonde (TVP), l'embolie pulmonaire, la thrombose des sinus veineux cérébraux, la thrombose veineuse sous-clavière, axillaire, la TVP de la veine cave inférieure et thrombose veineuse pelvienne.
d La pneumopathie interstitielle diffuse (PID)/pneumonie pour le cancer du sein précoce (CSP) inclut tous les termes préférentiels rapportés faisant partie des requêtes standardisées MedDRA (Standardised MedDRA Query SMQ pneumopathie interstitielle diffuse). Pour le cancer du sein métastatique (CSm), les termes préférentiels incluent pneumopathie interstitielle diffuse, pneumonie, pneumopathie organisée, fibrose pulmonaire et bronchiolite oblitérante.
e Considérés comme des EI uniquement en situation de CSm (MONARCH 2 et MONARCH 3).
f Considérés comme des EI uniquement en situation de CSP (monarchE).
g EI fréquents en situation de CSP (monarchE), EI très fréquents en situation de CSm (MONARCH 2 et MONARCH 3).
h EI fréquents en situation de CSm (MONARCH 2 et MONARCH 3), EI très fréquents en situation de CSP (monarchE).
Description d'effets indésirables sélectionnés
Neutropénie
Des neutropénies ont fréquemment été rapportées dans toutes les études. Dans l'étude monarchE, des neutropénies ont été rapportées chez 45,8 % des patients. Une diminution du nombre de neutrophiles à un grade 3 ou 4 (d'après les résultats des analyses biologiques) a été rapportée chez 19,1 % des patients traités par abémaciclib en association avec une hormonothérapie, avec un délai médian de survenue de 30 jours, et un délai médian de résolution de 16 jours. Une neutropénie fébrile a été rapportée chez 0,3 % des patients. Dans les études MONARCH 2 et MONARCH 3, des neutropénies ont été rapportées chez 45,1 % des patientes. Une diminution du nombre de neutrophiles à un grade 3 ou 4 a été rapportée chez 28,2 % des patientes traitées par abémaciclib en association avec des inhibiteurs de l'aromatase ou du fulvestrant (d'après les résultats des analyses biologiques). Le délai médian de survenue des neutropénies de grade 3 ou 4 était de 29 à 33 jours, et le délai médian de résolution était de 11 à 15 jours. Une neutropénie fébrile a été rapportée chez 0,9 % des patientes. Une modification de la dose est recommandée chez les patients chez lesquels survient une neutropénie de grade 3 ou 4 (voir rubrique Posologie et mode d'administration).
Diarrhée
La diarrhée a été l'effet indésirable le plus fréquemment rapporté (voir Tableau 8). L'incidence était plus élevée au cours du premier mois de traitement par abémaciclib et plus faible par la suite. Dans l'étude monarchE, le délai médian de survenue du premier épisode de diarrhée était de 8 jours quel que soit le grade. La durée médiane de la diarrhée était de 7 jours pour le grade 2 et de 5 jours pour le grade 3. Dans les études MONARCH 2 et MONARCH 3, le délai médian de survenue du premier épisode de diarrhée était d'environ 6 à 8 jours quel que soit le grade. La durée médiane de la diarrhée était de 9 à 12 jours pour le grade 2 et de 6 à 8 jours pour le grade 3. Le retour au grade initial de la diarrhée ou à un grade inférieur a été obtenu avec un traitement symptomatique tel que le lopéramide et/ou une adaptation posologique (voir rubrique Posologie et mode d'administration).
Transaminases augmentées
Dans l'étude monarchE, des élévations de l'ALAT et de l'ASAT ont été fréquemment rapportées (respectivement 12,3 % et 11,8 %) chez les patients traités par abémaciclib en association avec une hormonothérapie. Des élévations de grade 3 ou 4 de l'ALAT ou de l'ASAT (d'après les résultats des analyses biologiques) ont été rapportées chez 2,6 % et 1,6 % des patients. Le délai médian de survenue d'une élévation de grade 3 ou 4 de l'ALAT était de 118 jours, et le délai médian de résolution était de 14,5 jours. Le délai médian de survenue d'une élévation de grade 3 ou 4 de l'ASAT était de 90,5 jours, et le délai médian de résolution était de 11 jours. Dans les études MONARCH 2 et MONARCH 3, des élévations de l'ALAT et de l'ASAT ont été fréquemment rapportées (respectivement 15,1 % et 14,2 %) chez les patientes traitées par abémaciclib en association avec des inhibiteurs de l'aromatase ou du fulvestrant. Des élévations de grade 3 ou 4 de l'ALAT ou de l'ASAT (d'après les résultats des analyses biologiques) ont été rapportées respectivement chez 6,1 % et 4,2 % des patientes. Le délai médian de survenue d'une élévation de grade 3 ou 4 de l'ALAT était de 57 à 61 jours et le délai médian de résolution était de 14 jours. Le délai médian de survenue d'une élévation de grade 3 ou 4 de l'ASAT était de 71 à 185 jours, et le délai médian de résolution était de 13 à 15 jours. Une modification de la dose est recommandée chez les patients qui présentent une élévation de grade 3 ou 4 de l'ALAT ou de l'ASAT (voir rubrique Posologie et mode d'administration).
Créatinine
Bien qu'il ne s'agisse pas d'un effet indésirable, il a été mis en évidence que l'abémaciclib a entraîné une augmentation de la créatinine sérique. Dans l'étude monarchE, 99,3 % des patients ont présenté des élévations de la créatinine sérique (d'après les résultats des analyses biologiques) dont 0,5 % présentaient une élévation de grade 3 ou 4. Parmi les patients traités par une hormonothérapie seule, 91,0 % ont présenté une augmentation de la créatinine sérique (tous grades confondus). Dans les études MONARCH 2 et MONARCH 3, 98,3 % des patientes ont présenté des élévations de la créatinine sérique (d'après les résultats des analyses biologiques), dont 1,9 % des cas étaient de grade 3 ou 4. Chez les patientes traitées par un inhibiteur de l'aromatase ou du fulvestrant en monothérapie, 78,4 % ont rapporté une élévation de la créatinine sérique (tous grades confondus). Il a été démontré que l'abémaciclib entraîne une élévation de la créatinine sérique en raison de l'inhibition des transporteurs de la sécrétion tubulaire rénale sans affecter la fonction glomérulaire (mesurée par la clairance plasmatique de l'iohexol) (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Dans les études cliniques, les élévations de la créatinine sérique sont survenues au cours du premier mois de traitement par abémaciclib, sont restées élevées mais stables pendant toute la durée du traitement, ont été réversibles à l'arrêt du traitement, et n'étaient pas accompagnées de modifications des marqueurs de la fonction rénale tels que le taux d'azote uréique sanguin (BUN), la cystatine C, ou le débit de filtration glomérulaire calculé sur la base du dosage de la cystatine C.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT l'instauration du traitement, une numération des neutrophiles en valeur absolue (NAN) ≥ 1500/mm3, une numération plaquettaire ≥ 100 000/mm3 et un taux d'hémoglobine ≥ 8 g/dL sont recommandés.
SURVEILLANCE du traitement :
- Numération formule sanguine : avant l'initiation du traitement, toutes les deux semaines pendant les deux premiers mois, tous les mois pendant les deux mois suivants et selon les indications cliniques.
- ALAT (alanine aminotransférase) et ASAT (aspartate aminotransférase) : avant l'initiation du traitement, toutes les deux semaines pendant les deux premiers mois, tous les mois pendant les deux mois suivants et selon les indications cliniques.
- Signes et symptômes de thromboses veineuses profondes et d'embolies pulmonaires.
CONTACTER IMMEDIATEMENT LE MEDECIN en cas de :
- Symptômes tels que des frissons ou de la fièvre, toux, difficultés à respirer ou douleurs à la poitrine.
- Jambe gonflée et douloureuse, douleur dans la poitrine, essoufflement, respiration rapide ou fréquence cardiaque rapide.
- Diarrhée.
NE PAS consommer de jus de pamplemousse ni de pamplemousse durant le traitement.
NE PAS consommer de préparations à base de plantes contenant du millepertuis (Hypericum perforatum) durant le traitement.
Les femmes en âge de procréer doivent UTILISER une contraception efficace pendant le traitement et au moins 3 semaines apprès l'arrêt du traitement.PRUDENCE lors de la conduite de véhicules ou l'utilisation de machines ( risque de fatigue ou de sentations vertigineuses pendant le traitement).
EN CAS DE DIARRHEE, commencer, dès les premiers signes, un traitement avec des agents antidiarrhéiques, tels que le lopéramide. Boire beaucoup de liquides.
En cas de VOMISSEMENTS après avoir pris la dose ou en cas d'OUBLI d'une dose, prendre la dose suivante à l'heure habituelle.
Femmes en âge de procréer/Contraception féminine
Les femmes en âge de procréer doivent utiliser des méthodes de contraception hautement efficaces (par ex. une contraception à double-barrière) pendant le traitement et pendant au moins 3 semaines après l'arrêt du traitement (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Grossesse
Il n'existe aucune donnée concernant l'utilisation de l'abémaciclib chez la femme enceinte. Des études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Verzenios n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Allaitement
On ignore si l'abémaciclib est excrété dans le lait maternel. Un risque pour les nouveau- nés/nourrissons ne peut être exclu. Les patientes traitées par abémaciclib ne doivent pas allaiter.
Fertilité
L'effet de l'abémaciclib sur la fertilité humaine est inconnu. Bien qu'aucun effet sur la fertilité des mâles n'ait été observé chez les rats, les effets cytotoxiques observés sur l'appareil reproducteur mâle chez les souris, les rats, et les chiens, indiquent que l'abémaciclib est susceptible d'altérer la fertilité chez les mâles. Aucun effet indésirable sur les organes reproducteurs femelles chez les souris, les rates, ou les chiennes, ni aucun effet sur la fertilité des femelles et le développement embryonnaire précoce chez les rates n'a été observé (voir rubrique Données de sécurité préclinique).
Effets d'autres médicaments sur la pharmacocinétique de l'abémaciclib
L'abémaciclib est principalement métabolisé par le CYP3A4.
Inhibiteurs du CYP3A4
L'administration concomitante d'abémaciclib et d'inhibiteurs du CYP3A4 peut entraîner une augmentation des concentrations plasmatiques d'abémaciclib. Chez les patients atteints d'un cancer avancé et/ou métastatique, l'administration concomitante de clarithromycine, un inhibiteur du CYP3A4, a multiplié par 3,4 l'exposition plasmatique à l'abémaciclib et par 2,5 l'exposition plasmatique, avec une puissance ajustée, à l'abémaciclib libre combinée à ses métabolites actifs libres.
L'utilisation d'inhibiteurs puissants du CYP3A4 avec de l'abémaciclib doit être évitée. Si des inhibiteurs puissants du CYP3A4 doivent être administrés de façon concomitante, la dose d'abémaciclib doit être réduite (voir rubrique Posologie et mode d'administration), et une surveillance étroite de la survenue d'une toxicité doit être réalisée. Les exemples d'inhibiteurs puissants du CYP3A4 incluent (liste non exhaustive) : clarithromycine, itraconazole, kétoconazole, lopinavir/ritonavir, posaconazole ou voriconazole. Éviter de consommer du pamplemousse ou du jus de pamplemousse.
Aucun ajustement posologique n'est nécessaire pour les patients traités avec des inhibiteurs du CYP3A4 modérés ou faibles. Les signes de toxicités doivent cependant être surveillés de près.
Inducteurs du CYP3A4
L'administration concomitante d'abémaciclib et de rifampicine, un inducteur puissant du CYP3A4, a réduit de 95 % l'exposition plasmatique de l'abémaciclib et de 77 % l'exposition plasmatique, avec une puissance ajustée, à l'abémaciclib et ses métabolites actifs libres selon l'AUC0-8. L'utilisation concomitante d'inducteurs puissants du CYP3A4 (incluant [liste non exhaustive] : carbamazépine, phénytoïne, rifampicine et millepertuis) doit être évitée en raison du risque de diminution de l'efficacité de l'abémaciclib.
Effets de l'abémaciclib sur la pharmacocinétique d'autres médicaments
Médicaments substrats de transporteurs
L'abémaciclib et ses principaux métabolites actifs inhibent les transporteurs rénaux de cations organiques 2 (OCT2), les transporteurs MATE1 (multidrug and extrusion toxin protein) et MATE2-K.
Des interactions in vivo de l'abémaciclib avec des substrats cliniquement pertinents de ces transporteurs, tels que le dofétilide ou la créatinine, peuvent se produire (voir rubrique Effets indésirables). Dans une étude clinique d'interactions médicamenteuses avec de la metformine (substrat d'OCT2, MATE1 et 2) administrée concomitamment à 400 mg d'abémaciclib, une faible augmentation mais cliniquement non pertinente (37 %) de l'exposition plasmatique à la metformine a été observée. Il s'est avéré que cela était dû à une réduction de la sécrétion rénale avec une filtration glomérulaire non affectée.
Chez des sujets sains, l'administration concomitante d'abémaciclib et de lopéramide, un substrat de la glycoprotéine-P (P-gp), a entraîné une augmentation de l'exposition plasmatique au lopéramide de9 % selon l'AUC 0-8 et de 35 % selon la Cmax. Cela n'a pas été considéré comme cliniquement pertinent. Toutefois, compte tenu de l'inhibition in vitro de la P-gp et de la protéine de résistance au cancer du sein (breast cancer resistance protein, BCRP) observée avec l'abémaciclib, des interactions in vivo de l'abémaciclib avec des substrats de ces transporteurs à marge thérapeutique étroite comme la digoxine ou le dabigatran etexilate sont possibles.
Lors d'une étude clinique chez des patientes atteintes d'un cancer du sein, il n'y a pas eu d'interaction pharmacocinétique cliniquement pertinente entre l'abémaciclib et l'anastrozole, le fulvestrant, l'exémestane, le létrozole ou le tamoxifène.
On ignore à l'heure actuelle si l'abémaciclib pourrait réduire l'efficacité des contraceptifs hormonaux à action systémique.
Le traitement par Verzenios doit être instauré et suivi par des médecins expérimentés dans l'utilisation des traitements anticancéreux.
Posologie
Verzenios en association avec une hormonothérapie
La dose recommandée d'abémaciclib est de 150 mg deux fois par jour lorsqu'il est administré en association avec une hormonothérapie. Se référer au Résumé des caractéristiques du produit de l'hormonothérapie co-administrée pour en connaître la posologie recommandée.
Durée du traitement
Cancer du sein précoce
Verzenios doit être pris de manière continue pendant deux ans, ou jusqu'à la rechute de la maladie ou la survenue d'une toxicité inacceptable.
Cancer du sein avancé ou métastatique
Verzenios doit être pris de manière continue tant qu'un bénéfice clinique est observé chez la patiente ou jusqu'à la survenue d'une toxicité inacceptable.
En cas de vomissements ou d'omission d'une dose de Verzenios, le patient doit être informé de prendre la dose suivante à l'heure prévue ; une dose supplémentaire ne doit pas être prise.
Adaptations posologiques
La prise en charge de certains effets indésirables peut nécessiter une interruption du traitement et/ou une réduction posologique, comme indiqué dans les Tableaux 1-7.
Tableau 1. Adaptations posologiques recommandées en cas d'effets indésirables
| | Dose de Verzenios en association |
| Dose recommandée | 150 mg deux fois par jour |
| Première réduction posologique | 100 mg deux fois par jour |
| Deuxième réduction posologique | 50 mg deux fois par jour |
Tableau 2. Recommandations de prise en charge des toxicités hématologiques
La numération formule sanguine doit être surveillée avant l'initiation du traitement par Verzenios, toutes les deux semaines pendant les deux premiers mois, tous les mois pendant les deux mois suivants et selon les indications cliniques. Avant l'instauration du traitement, une numération des neutrophiles en valeur absolue (NAN) ≥ 1 500/mm3, une numération plaquettaire ≥ 100 000/mm3 et un taux d'hémoglobine ≥ 8 g/dL sont recommandés.
| Toxicitéa,b | Recommandations de prise en charge |
| Grade 1 ou 2 | Aucune adaptation posologique n'est requise. |
| Grade 3 | Interrompre le traitement jusqu'au retour à une toxicité de grade ≤ 2 Aucune réduction de dose n'est requise. |
| Grade 3, récurrente ; ou grade 4 | Interrompre le traitement jusqu'au retour à une toxicité de grade ≤ 2 Reprendre le traitement à la dose immédiatement inférieure. |
| Le patient nécessite l'administration de facteurs de croissance hématopoïétiques | Interrompre le traitement par abémaciclib pendant au moins 48 heures après l'administration de la dernière dose de facteurs de croissance hématopoïétiques et jusqu'au retour à une toxicité de grade ≤ 2. Reprendre le traitement à la dose immédiatement inférieure sauf si la dose a déjà été réduite en raison de la toxicité ayant conduit à l'administration du facteur de croissance. |
a Critères Communs de Toxicité pour les évènements indésirables (CTCAE) du National Cancer Institute (NCI)
b NAN : Grade 1 : NAN < LIN à 1 500/mm3 ; Grade 2 : NAN 1 000 à < 1 500/mm3; Grade 3 : NAN 500 à < 1 000/mm3 ; Grade 4 : NAN < 500/mm3
LIN = Limite Inférieure de la Normale
Tableau 3. Recommandations de prise en charge de la diarrhée
Un traitement avec des agents antidiarrhéiques, tels que le lopéramide, doit être instauré au premier signe de selles molles.
| Toxicité a | Recommandations de prise en charge |
| Grade 1 | Aucune adaptation posologique requise. |
| Grade 2 | Si aucun retour à une toxicité de grade ≤ 1 n'est obtenu dans les 24 heures, interrompre le traitement jusqu'à disparition de la toxicité. Aucune réduction de dose n'est requise. |
| Persistance ou récurrence d'une toxicité de grade 2 après réintroduction de la même dose malgré la mise en place de mesures symptomatiques optimales | Interrompre le traitement jusqu'à un retour à une toxicité de grade ≤ 1. Reprendre le traitement à la dose immédiatement inférieure. |
| Grade 3 ou 4 ou qui impose une hospitalisation |
a Critères Communs de Toxicité pour les évènements indésirables (CTCAE) du National Cancer Institute (NCI)
Tableau 4. Recommandations de prise en charge des élévations des transaminases
L'ALAT (alanine aminotransférase) et l'ASAT (aspartate aminotransférase) doivent être surveillées avant l'initiation du traitement par Verzenios, toutes les deux semaines pendant les deux premiers mois, tous les mois pendant les deux mois suivants et selon les indications cliniques.
| Toxicité a | Recommandations de prise en charge |
| Grade 1 (> LSN à 3,0 x LSN) Grade 2 (> 3,0 à 5,0 x LSN) | Aucune adaptation posologique requise. |
| Persistance ou récurrence d'une toxicité de grade 2, ou grade 3 (> 5,0 à 20,0 x LSN) | Interrompre le traitement jusqu'au retour à une toxicité du niveau de l'inclusion ou au Grade 1. Reprendre le traitement à la dose immédiatement inférieure. |
| Elévation des ASAT et/ou ALAT > 3 x LSN avec bilirubine totale > 2 x LSN, en l'absence de cholestase | Arrêter l'abémaciclib. |
| Grade 4 (> 20,0 x LSN) | Arrêter l'abémaciclib. |
a Critères Communs de Toxicité pour les évènements indésirables (CTCAE) du National Cancer Institute (NCI)
LSN = Limite supérieure de la normale
Tableau 5. Recommandations de prise en charge d'une pneumopathie interstitielle diffuse (PID)/pneumonie
| Toxicité a | Recommandations de prise en charge |
| Grade 1 ou 2 | Aucune adaptation posologique n'est requise. |
| Persistance ou récurrence d'une toxicité de grade 2 sans retour à l'état initial ou à un grade 1 dans les 7 jours malgré la mise en place de mesures symptomatiques optimales | Interrompre le traitement jusqu'au retour à une toxicité du niveau de l'inclusion ou au Grade 1. Reprendre le traitement à la dose immédiatement inférieure. |
| Grade 3 ou 4 | Arrêter l'abémaciclib. |
a Critères Communs de Toxicité pour les évènements indésirables (CTCAE) du National Cancer Institute (NCI)
Tableau 6. Recommandations de prise en charge des évènements thrombo-emboliques veineux
| Toxicité a | Recommandations de prise en charge |
| Cancer du sein précoce | |
| Tous grades (1, 2, 3, ou 4) | Interrompre le traitement et traiter en fonction du tableau clinique. L'abémaciclib peut être repris lorsque l'état clinique du patient est stable. |
| Cancer du sein avancé ou métastatique | |
| Grade 1 ou 2 | Aucune modification de la posologie n'est requise. |
| Grade 3 ou 4 | Interrompre le traitement et traiter en fonction du tableau clinique. L'abémaciclib peut être repris lorsque l'état clinique de la patiente est stable. |
a Critères Communs de Toxicité pour les évènements indésirables (CTCAE) du National Cancer Institute (NCI)
Tableau 7. Recommandations de prise en charge des toxicités non-hématologiques (à l'exclusion de la diarrhée, des élévations des transaminases, d'une PID/pneumonie et des évènements thrombo-emboliques veineux)
| Toxicité a | Recommandations de prise en charge |
| Grade 1 ou 2 | Aucune adaptation posologique n'est requise. |
| Persistance ou | |
| récurrence d'une | |
| toxicité de grade 2 sans | |
| retour à la valeur | |
| initiale ou à un grade 1 | Interrompre le traitement jusqu'au retour à une toxicité de grade ≤ 1 |
| dans les 7 jours malgré | Reprendre le traitement à la dose immédiatement inférieure. |
| la mise en place de | |
| mesures | |
| symptomatiques | |
| optimales | |
| Grade 3 ou 4 | |
a Critères Communs de Toxicité pour les évènements indésirables (CTCAE) du National Cancer Institute (NCI)
Inhibiteurs du CYP3A4
L'utilisation concomitante d'inhibiteurs puissants du CYP3A4 doit être évitée. Si l'utilisation d'un inhibiteur puissant du CYP3A4 ne peut être évitée, la dose d'abémaciclib doit être réduite à 100 mg deux fois par jour.
Chez les patients qui ont eu une dose réduite à 100 mg d'abémaciclib deux fois par jour et chez lesquels l'administration concomitante d'un inhibiteur puissant du CYP3A4 ne peut être évitée, la dose d'abémaciclib doit être davantage réduite à 50 mg deux fois par jour.
Chez les patients qui ont eu une dose réduite à 50 mg d'abémaciclib deux fois par jour et chez lesquels l'administration concomitante d'un inhibiteur puissant du CYP3A4 ne peut être évitée, la dose d'abémaciclib peut être maintenue avec une surveillance étroite de signes de toxicité. Autrement, la dose d'abémaciclib peut être davantage réduite à 50 mg une fois par jour ou le traitement peut être interrompu.
Si l'inhibiteur du CYP3A4 est interrompu, la dose d'abémaciclib doit être ré-augmentée à la dose utilisée avant l'initiation de l'inhibiteur du CYP3A4 (après 3 à 5 demi-vies de l'inhibiteur du CYP3A4).
Populations particulières
Sujets âgés
Aucune adaptation posologique n'est requise en fonction de l'âge (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée. Il n'existe pas de données concernant l'administration d'abémaciclib à des patients présentant une insuffisance rénale sévère, une néphropathie en phase terminale, ou chez les patients sous dialyse (voir rubrique Propriétés pharmacocinétiques). L'abémaciclib doit être administré avec prudence chez les patients présentant une insuffisance rénale sévère avec une surveillance étroite des signes de toxicité.
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère (Child Pugh A) ou modérée (Child Pugh B). Chez les patients présentant une insuffisance hépatique sévère (Child Pugh C), il est recommandé de diminuer la fréquence de prise à une fois par jour (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité d'abémaciclib chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas été établies.
Aucune donnée n'est disponible.
Mode d'administration
Verzenios est à prendre par voie orale.
La dose peut être prise avec ou sans nourriture. Elle ne doit pas être prise avec du pamplemousse ou du jus de pamplemousse (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Les patients doivent prendre les doses approximativement aux mêmes heures chaque jour.
Le comprimé doit être avalé en entier (les patients ne doivent pas le mâcher, l'écraser ni le couper avant de l'avaler).
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de conditions particulières de conservation.
Sans objet.
En cas de surdosage d'abémaciclib, une fatigue et une diarrhée peuvent survenir. Une prise en charge globale symptomatique devra être réalisée.
Classe pharmacothérapeutique : Agents antinéoplasiques, inhibiteurs de protéines kinases, Code ATC : L01EF03
Mécanisme d'action
L'abémaciclib est un inhibiteur puissant et sélectif des kinases cycline-dépendantes 4 et 6 (CDK4 et CDK6), et plus particulièrement envers le complexe cycline D1/CDK4 dans les dosages enzymatiques. L'abémaciclib empêche la phosphorylation de la protéine du rétinoblastome (Rb), en bloquant la progression du cycle cellulaire de la phase G1 à la phase S lors de la division cellulaire, ce qui entraîne
l'arrêt de la croissance tumorale. Dans les lignées cellulaires de cancer du sein à récepteurs œstrogéniques positifs, l'inhibition ciblée durable obtenue avec l'abémaciclib a empêché le rebond de la phosphorylation de Rb, ce qui s'est traduit par une sénescence cellulaire et une apoptose. In vitro, les cellules cancéreuses Rb négatives et déplétées en Rb sont généralement moins sensibles à l'abémaciclib. Dans les modèles de xénogreffe de cancer du sein, l'abémaciclib administré quotidiennement sans interruption à des concentrations cliniquement pertinentes, associé ou non avec des anti-œstrogènes a permis d'obtenir une réduction de la taille de la tumeur.
Effets pharmacodynamiques
Chez les patients atteints d'un cancer, l'abémaciclib inhibe CDK4 et CDK6 comme le montre l'inhibition de la phosphorylation de Rb et de la topoisomérase II alpha, ce qui entraîne une inhibition du cycle cellulaire en amont du point de restriction G1.
Électrophysiologie cardiaque
L'effet de l'abémaciclib sur l'intervalle QTcF (corrigé selon la méthode Fridericia) a été évalué chez 144 patientes atteintes d'un cancer avancé. Aucune modification importante (c'est-à-dire > 20 ms) de l'intervalle QTcF n'a été détectée à la concentration moyenne maximale à l'état d'équilibre d'abémaciclib, observée à la suite d'un schéma posologique thérapeutique.
Dans une analyse de la relation exposition-réponse chez des sujets sains à des expositions comparables à une dose de 200 mg deux fois par jour, l'abémaciclib n'a pas entraîné d'allongement de l'intervalle QTcF de façon cliniquement significative.
Efficacité et sécurité cliniques
Cancer du sein précoce
Étude de phase III randomisée monarchE : Verzenios en association avec une hormonothérapie L'efficacité et la sécurité de Verzenios en association avec une hormonothérapie adjuvante ont été évaluées dans monarchE, une étude de phase III randomisée, en ouvert, à deux cohortes, menée chez des femmes et des hommes atteints d'un cancer du sein précoce HR-positif, HER2-négatif, avec atteinte ganglionnaire et haut risque de rechute. Dans la Cohorte 1, le haut risque de rechute était défini par les caractéristiques cliniques et histopathologiques suivantes : soit ≥ 4 pALN (ganglions lymphatiques axillaires positifs) soit 1-3 pALN et au moins l'un des critères suivants : taille de la tumeur ≥ 5 cm ou de grade histologique 3.
Au total, 5 637 patients ont été randomisés selon un rapport 1:1 pour recevoir, pendant 2 ans, Verzenios 150 mg deux fois par jour associé à une hormonothérapie standard au choix du médecin, ou pour recevoir une hormonothérapie standard seule. La randomisation a été stratifiée selon la chimiothérapie antérieure, le statut de ménopause et la région. Les hommes ont été stratifiés comme post-ménopausés. Les patients avaient terminé un traitement locorégional définitif (avec ou sans chimiothérapie néo-adjuvante ou adjuvante). Les patients devaient être rétablis des effets secondaires aigus de toute chimiothérapie ou radiothérapie antérieures. Une période de sevrage thérapeutique de 21 jours après la chimiothérapie et de 14 jours après la radiothérapie était obligatoire avant la randomisation. Les patients étaient autorisés à recevoir jusqu'à 12 semaines d'hormonothérapie adjuvante avant la randomisation. Le traitement adjuvant par fulvestrant n'était pas autorisé comme hormonothérapie standard. Les patients présentant un indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 ou 1 étaient éligibles. Les patients présentant un antécédent de thrombo-embolie veineuse étaient exclus de l'étude. Après la fin de la période de traitement de l'étude, les patients des deux bras de traitement ont continué à être traités par hormonothérapie adjuvante pendant une durée cumulative d'au moins 5 ans, et jusqu'à 10 ans, si médicalement approprié. Des agonistes de la LHRH étaient administrés quand cliniquement nécessaire aux femmes pré- et périménopausées, et aux hommes.
Parmi les 5 637 patients randomisés, 5 120 étaient inclus dans la Cohorte 1, représentant 91 % de la population en intention de traiter. Dans la cohorte 1, les données démographiques des patients et les caractéristiques tumorales à l'état initial étaient réparties de façon équilibrée dans les bras de traitement. L'âge médian des patients inclus était d'environ 51 ans (intervalle de 22 à 89 ans) ; 15 % des patients avaient 65 ans ou plus, 99 % étaient des femmes, 71 % étaient de type caucasien, 24 % étaient d'origine ethnique asiatique et 5 % d'autres groupes ethniques. Quarante-trois pour cent des patientes étaient pré- ou périménopausées. La plupart des patients avaient reçu une chimiothérapie antérieure (36 % néoadjuvante, 62 % adjuvante) et une radiothérapie antérieure (96 %).
L'hormonothérapie initiale reçue par les patients incluait létrozole (39 %), tamoxifène (31 %), anastrozole (22 %) ou exémestane (8 %).
Soixante-cinq pour cent des patients présentaient 4 ganglions lymphatiques positifs ou plus, 41 % avaient une tumeur de grade 3 et 24 % présentaient une tumeur de taille ≥ 5 cm lors de la chirurgie.
Dans la population en Intention de Traiter (ITT), le critère principal d'évaluation était la survie sans maladie invasive (Invasive Disease-Free Survival ou IDFS), définie comme le temps entre la date de randomisation et la date de première survenue d'une récidive ipsilatérale d'un cancer du sein invasif, d'une récidive régionale d'un cancer du sein invasif, d'une récidive à distance, d'un cancer du sein invasif controlatéral, d'un second cancer invasif primitif autre qu'un cancer du sein, ou d'un décès quelle qu'en soit la cause. Dans la population en ITT, le principal critère secondaire d'évaluation était la survie sans récidive à distance (Distant Relapse Free Survival ou DRFS), définie comme le temps entre la date de randomisation et la date de première survenue d'une récidive à distance, ou d'un décès quelle qu'en soit la cause.
L'objectif principal de l'étude a été atteint lors de l'analyse intermédiaire pré-planifiée (à la date du 16 mars 2020). Une amélioration statistiquement significative de l'IDFS a été observée chez les patients ayant reçu Verzenios associé à une hormonothérapie versus une hormonothérapie seule dans la population en ITT. L'approbation a été octroyée pour la grande sous-population, la Cohorte 1.
Dans une analyse supplémentaire (à la date du 01 avril 2021), 91 % des patients de la Cohorte 1 étaient sortis de la période de traitement de l'étude de 2 ans et la durée médiane du suivi était de 27,7 mois.
Les résultats d'efficacité dans la Cohorte 1 sont résumés dans le Tableau 9 et la Figure 1.
Tableau 9. Étude monarchE : Résumé des données d'efficacité (population de la Cohorte 1)
| | Verzenios plus hormonothérapie N = 2 555 | Hormonothérapie seule N = 2 565 |
| Survie sans maladie invasive (Invasive Disease-Free Survival ou IDFS) | | |
| Nombre de patients avec évènement (n, %) | 218 (8,5) | 318 (12,4) |
| Hazard ratio (IC à 95 %) | 0,680 (0,572 ; 0,808) | |
| IDFS à 24 mois (%, IC à 95 %) | 92,6 (91,4 ; 93,5) | 89,6 (88,3 ; 90,8) |
| Survie sans récidive à distance (Distant Relapse Free Survival ou DRFS) | | |
| Nombre de patients avec évènement (n, %) | 179 (7,0) | 266 (10,4) |
| Hazard ratio (IC à 95 %) | 0,669 (0,554 ; 0,809) | |
| DRFS à 24 mois (%, IC à 95 %) | 94,1 (93,0 ; 95,0) | 91,2 (90,0 ; 92,3) |
Abréviations : IC = intervalle de confiance Données à la date du : 01 Avril 2021
Figure 1. Étude monarchE : Courbe de Kaplan-Meier de l'IDFS (Évaluation par l'investigateur, population de la Cohorte 1)
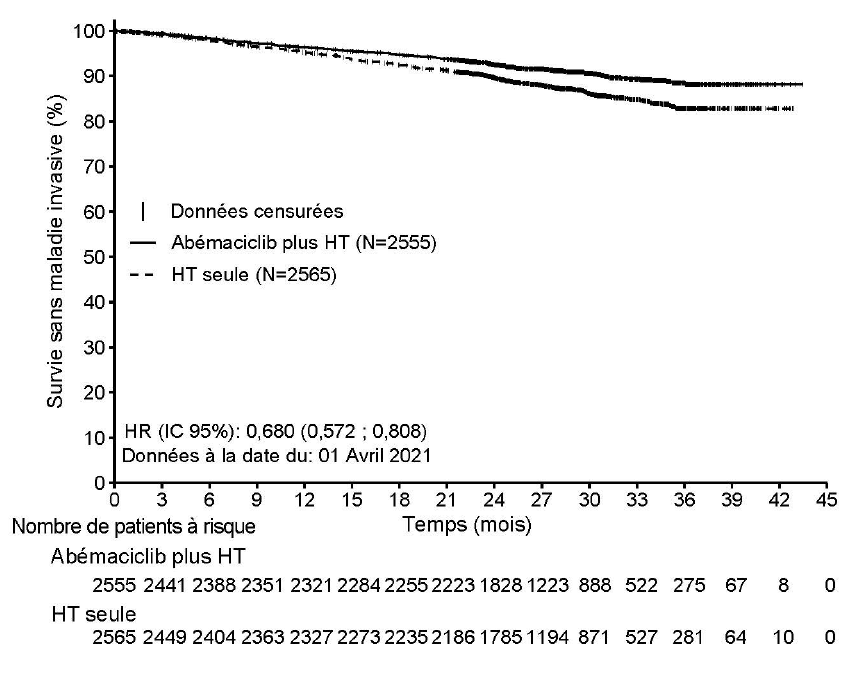
Abréviations : IC = intervalle de confiance, HT = hormonothérapie, HR = Hazard Ratio, IDFS (Invasive Disease-Free Survival) = survie sans maladie invasive, N = nombre de patients dans la population.
Données à la date du : 01 Avril 2021
Le bénéfice a été observé dans les sous-groupes de patients définis par la région géographique, le statut ménopausique et la chimiothérapie antérieure au sein de la Cohorte 1.
Cancer du sein avancé ou métastatique
Étude de phase III randomisée MONARCH 3 : Verzenios en association avec des inhibiteurs de l'aromatase
L'efficacité et la sécurité de Verzenios en association avec un inhibiteur de l'aromatase (anastrozole ou létrozole) ont été évaluées dans MONARCH 3, une étude de phase III randomisée, en double aveugle, contrôlée versus placebo, menée chez des femmes atteintes d'un cancer du sein RH+, HER2- localement avancé ou métastatique n'ayant pas reçu de traitement systémique antérieur pour ce stade de leur maladie. Les patientes ont été randomisées selon un rapport 2:1 pour recevoir Verzenios 150 mg deux fois par jour associé à un inhibiteur de l'aromatase non stéroïdien administré quotidiennement à la dose recommandée versus le placebo en association à un inhibiteur de l'aromatase non stéroïdien avec le même schéma posologique. Le critère principal était la Survie Sans Progression (SSP) évaluée par l'investigateur selon les critères RECIST 1.1 ; les principaux critères secondaires d'efficacité étaient le taux de réponse objective (TRO), le taux de bénéfice clinique (TBC) et la survie globale (SG).
L'âge médian des patientes incluses dans l'étude était de 63 ans (intervalle de 32 à 88 ans). Environ 39 % des patientes avaient été traitées par chimiothérapie et 44 % avaient reçu un traitement antihormonal dans le cadre d'un traitement (néo)adjuvant. Les patientes ayant été traitées par hormonothérapie (néo)adjuvante devaient avoir terminé ce traitement depuis au moins 12 mois avant leur randomisation dans l'étude. La majorité des patientes (96 %) présentaient une maladie métastatique à l'inclusion. Environ 22 % des patientes avaient des atteintes exclusivement osseuses et 53 % des patientes avaient des métastases viscérales.
L'étude a atteint son critère principal d'amélioration de la SSP. Les principaux résultats d'efficacité sont résumés dans le Tableau 10 et la Figure 2.
Tableau 10. Étude MONARCH 3 : Résumé des données d'efficacité (évaluation de l'investigateur, population en intention de traiter)
| | Verzenios plus inhibiteur de l'aromatase | Placebo plus inhibiteur de l'aromatase |
| Survie sans progression | N = 328 | N = 165 |
| Évaluation par l'investigateur, nombre d'événements (%) | 138 (42,1) | 108 (65,5) |
| Médiane [mois] (IC à 95 %) | 28,18 (23,51 ; NA) | 14,76 (11,24 ; 19,20) |
| Hazard ratio (IC à 95 %) et valeur de p | 0,540 (0,418 ; 0,698), p = 0,000002 | |
| Revue radiographique indépendante, nombre d'événements (%) | 91 (27,7) | 73 (44,2) |
| Médiane [mois] (IC à 95 %) | NA (NA ; NA) | 19,36 (16,37 ; 27,91) |
| Hazard ratio (IC à 95 %) et valeur de p | 0,465 (0,339 ; 0,636); p < 0,000001 | |
| Taux de réponse objectiveb [%] (IC à 95 %) | 49,7 (44,3 ; 55,1) | 37,0 (29,6 ; 44,3) |
| Durée de la réponse [mois] (IC à 95 %) | 27,39 (25,74 ; NA) | 17,46 (11,21 ; 22,19) |
| Réponse objective des patientes avec une maladie mesurablea | N = 267 | N = 132 |
| Taux de réponse objectiveb [%] (IC à 95 %) | 61,0 (55,2 ; 66,9) | 45,5 (37,0 ; 53,9) |
| Réponse complète (%) | 3,4 | 0 |
| Réponse partielle (%) | 57,7 | 45,5 |
| Taux de bénéfice cliniquec (maladie mesurable) [%] (IC à 95 %) | 79,0 (74,1 ; 83,9) | 69,7 (61,9 ; 77,5) |
a Maladie mesurable définie selon les critères RECIST version 1.1
b Réponse complète + réponse partielle
c Réponse complète + réponse partielle + maladie stable pendant ≥ 6 mois N = nombre de patientes ; IC = intervalle de confiance ; NA = non atteint.
Figure 2. Étude MONARCH 3 : Courbe de Kaplan-Meier de la survie sans progression (Évaluation par l'investigateur, population en intention de traiter)
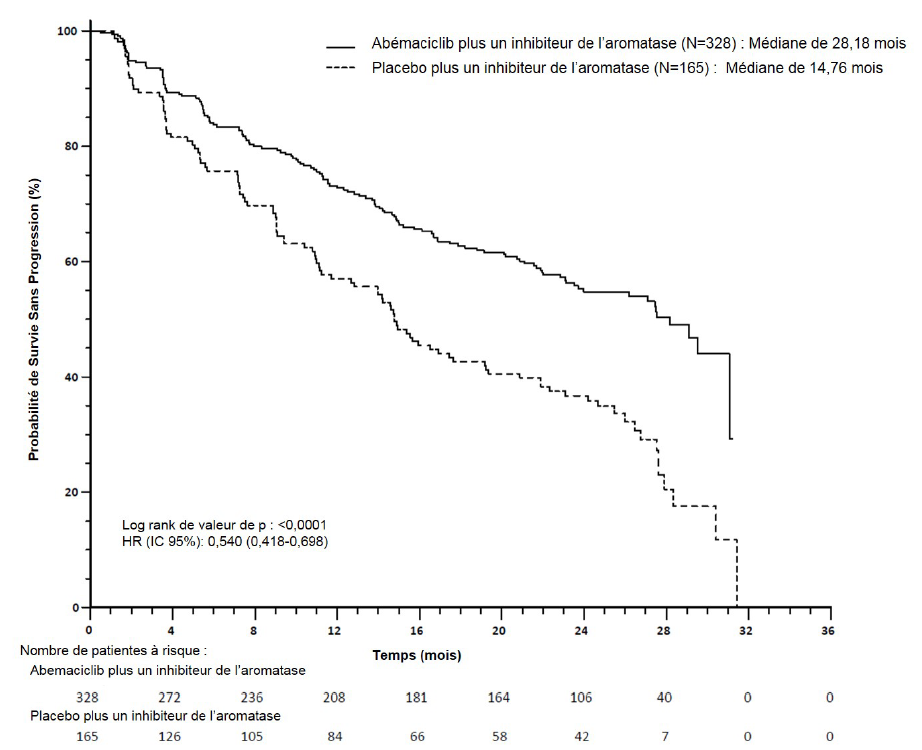
La survie sans progression (SSP) a été significativement prolongée dans le bras Verzenios plus inhibiteur de l'aromatase (AI), (HR de 0,540 [IC à 95 % : 0,418 ; 0,698]) ; la SSP médiane était de 28,18 mois dans le bras Verzenios plus AI et de 14,76 mois dans le bras placebo plus AI. Ces résultats correspondent à une réduction cliniquement significative du risque de progression de la maladie ou de décès de 46 % chez les patientes traitées par l'association d'abémaciclib avec un inhibiteur de l'aromatase.
Les données de survie globale n'étaient pas matures au moment de l'analyse finale de la SSP (93 événements observés dans les deux bras). Le HR était de 1,057 (IC à 95 % : 0,683 ; 1,633), p = 0,8017.
Une série d'analyses en sous-groupes préspécifiés de la SSP a montré des résultats concordants dans tous les sous-groupes de patientes par : âge (< 65 ans ou ≥ 65 ans), site de la maladie, stade de la maladie (métastatique de novo versus métastatique récurrente versus récurrente localement avancée), présence d'une maladie mesurable, statut des récepteurs à la progestérone, et indice de performance ECOG à l'inclusion. Une réduction du risque de progression de la maladie ou de décès a été observée chez les patientes présentant une maladie viscérale (HR de 0,567 [IC à 95 % : 0,407 ; 0,789]), médiane de SSP : 21,6 mois versus 14,0 mois ; chez les patientes présentant des atteintes exclusivement osseuses (HR de 0,565 [IC à 95 % : 0,306 ; 1,044]) ; et chez les patientes présentant une maladie mesurable (HR de 0,517 [IC à 95 % : 0,392 ; 0,681]).
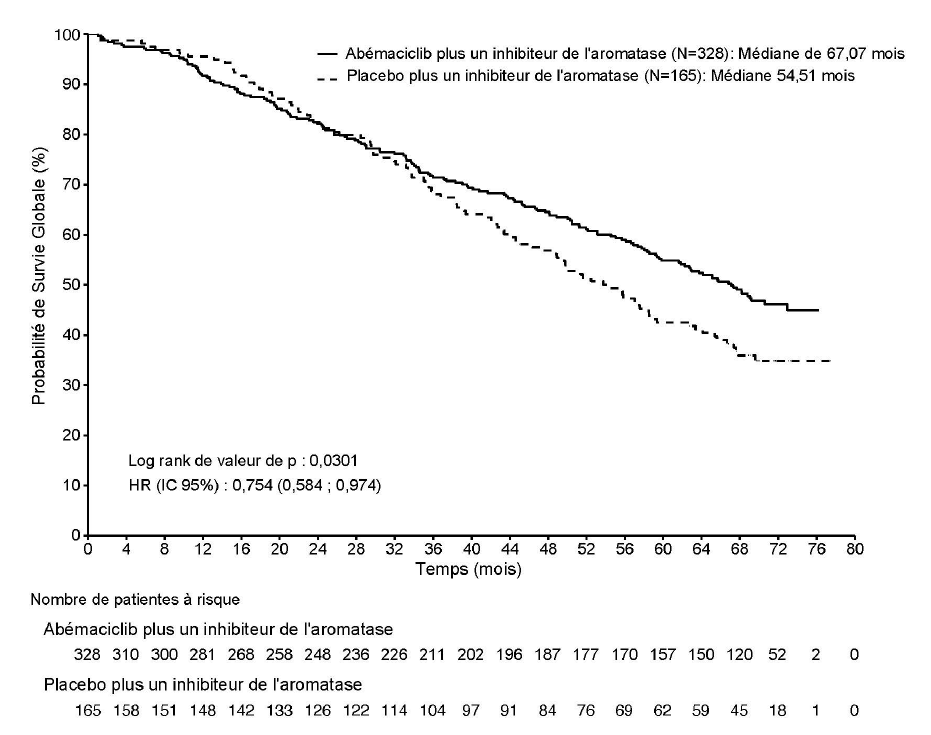
Lors de la première analyse intermédiaire de la SG, 197 événements ont été observés dans les deux bras, et le HR était de 0,786 (IC 95 % : 0,589 ; 1,049).
Lors de la deuxième analyse intermédiaire de la SG, 255 événements ont été observés dans les deux bras. La SG médiane était de 67,1 mois dans le bras abémaciclib plus AI et de 54,5 mois dans le bras placebo plus AI. Etant donné que le HR observé de 0,754 (IC 95 % : 0,584 ; 0,974) n'a pas atteint la significativité statistique (Figure 3), l'étude continue afin de caractériser complètement la survie globale.
Figure 3. Étude MONARCH 3 : Courbe de Kaplan-Meier de la survie globale (Population en intention de traiter)
Étude de phase III randomisée MONARCH 2 : Verzenios en association avec le fulvestrant
L'efficacité et la sécurité de Verzenios en association avec le fulvestrant ont été évaluées dans MONARCH 2, une étude de phase III randomisée, en double aveugle, contrôlée versus placebo, menée chez des femmes atteintes d'un cancer du sein RH+, HER2- localement avancé ou métastatique. Les patientes ont été randomisées selon un rapport 2:1 pour recevoir Verzenios 150 mg deux fois par jour associé à du fulvestrant 500 mg à intervalles de 1 mois, avec une dose supplémentaire de 500 mg administrée deux semaines après la dose initiale, versus placebo associé à du fulvestrant selon le même schéma posologique. Le critère principal était la SSP évaluée par l'investigateur selon les critères RECIST 1.1 ; les principaux critères secondaires d'efficacité étaient le taux de réponse objective (TRO), le taux de bénéfice clinique (TBC) et la survie globale (SG).
L'âge médian des patientes incluses était de 60 ans (intervalle de 32 à 91 ans). Dans chaque bras de traitement, la majorité des patientes étaient de type caucasien, et n'avaient pas antérieurement été traitées par chimiothérapie pour leur maladie métastatique. 17 % des patientes étaient en pré/périménopause en suppression ovarienne avec un agoniste de la LH-RH. Environ 56 % des patientes présentaient des métastases viscérales. Environ 25 % des patientes présentaient une hormonorésistance primaire (progression sous hormonothérapie dans les 2 premières années d'hormonothérapie adjuvante ou dans les 6 premiers mois d'une hormonothérapie en première ligne pour un cancer du sein métastatique), et, pour la majorité, une hormonorésistance développée plus
tard. 59 % des patientes avaient une hormonothérapie plus récente dans le cadre d'un traitement (néo)adjuvant, et 38 % dans un contexte métastatique.
L'étude a atteint son critère principal d'amélioration de la SSP. Les principaux résultats d'efficacité sont résumés dans le Tableau 11 et la Figure 4.
Tableau 11. Étude MONARCH 2 : Résumé des données d'efficacité (Évaluation par l'investigateur, population en intention de traiter)
| | Verzenios plus fulvestrant | Placebo plus fulvestrant |
| Survie sans progression | N = 446 | N = 223 |
| Évaluation par l'investigateur, nombre d'événements (%) | 222 (49,8) | 157 (70,4) |
| Médiane [mois] (IC à 95 %) | 16,4 (14,4 ; 19,3) | 9,3 (7,4 ; 12,7) |
| Hazard ratio (IC à 95 %) et valeur de p | 0,553 (0,449 ; 0,681), p = 0,0000001 | |
| Revue radiographique indépendante, nombre d'événements (%) | 164 (36,8) | 124 (55,6) |
| Médiane [mois] (IC à 95 %) | 22,4 (18,3 ; NA) | 10,2 (5,8 ; 14,0) |
| Hazard ratio (IC à 95 %) et valeur de p | 0,460 (0,363 ; 0,584) ; p < 0,000001 | |
| Taux de réponse objectiveb[%] (IC à 95 %) | 35,2 (30,8 ; 39,6) | 16,1 (11,3 ; 21,0) |
| Durée de la réponse [mois] (IC à 95 %) | NA (18,05 ; NA) | 25,6 (11,9 ; 25,6) |
| Réponse objective des patientes ayant une maladie mesurablea | N = 318 | N = 164 |
| Taux de réponse objectiveb [%] (IC à 95 %) | 48,1 (42,6 ; 53,6) | 21,3 (15,1 ; 27,6) |
| Réponse complète (%) | 3,5 | 0 |
| Réponse partielle (%) | 44,7 | 21,3 |
| Taux de bénéfice cliniquec (maladie mesurable) [%] (IC à 95 %) | 73,3 (68,4 ; 78,1) | 51,8 (44,2 ; 59,5) |
a Maladie mesurable définie selon les critères RECIST version 1.1
b Réponse complète + réponse partielle
c Réponse complète + réponse partielle + maladie stable pendant ≥ 6 mois N = nombre de patientes ; IC = intervalle de confiance ; NA = non atteint.
Figure 4. Étude MONARCH 2 : Courbe de Kaplan-Meier de la survie sans progression (Évaluation par l'investigateur, population en intention de traiter)
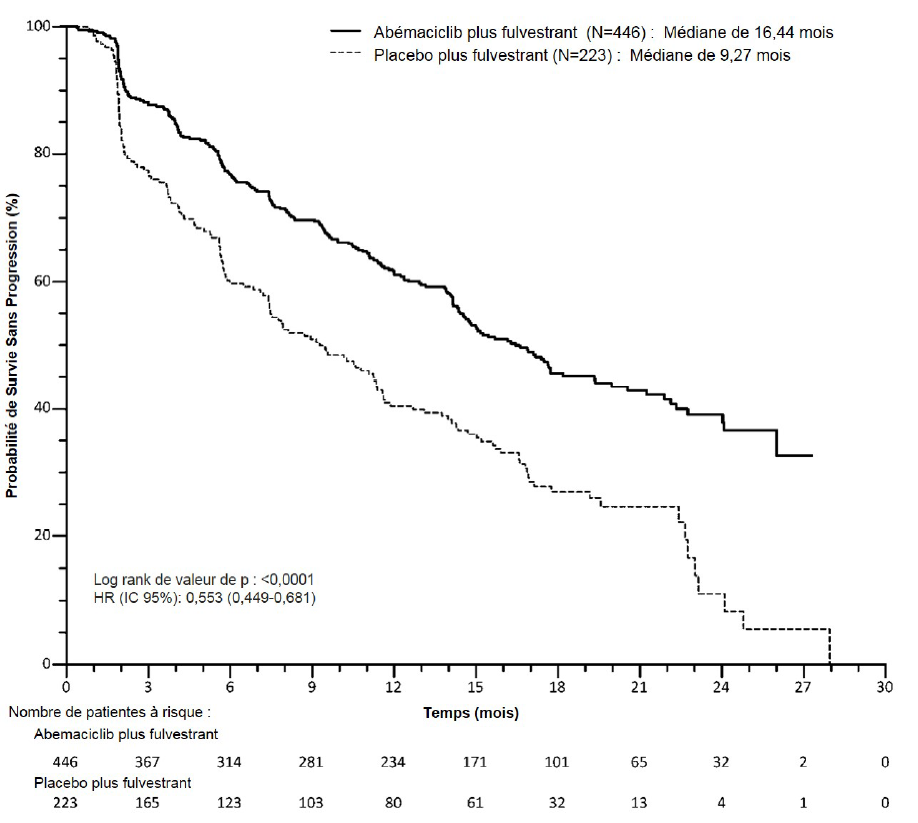
La SSP médiane a été prolongée de manière significative dans le bras Verzenios plus fulvestrant (HR de 0,553 [IC à 95 % : 0,449 ; 0,681]). La SSP médiane était de 16,4 mois versus 9,3 mois dans le bras placebo plus fulvestrant. Ces résultats correspondent à une réduction cliniquement significative de 44,7 % du risque de progression de la maladie ou de décès et à une amélioration de 7,2 mois de la SSP médiane chez les patientes traitées par Verzenios associé à du fulvestrant. L'association de Verzenios avec le fulvestrant a permis de prolonger la survie sans progression, sans impact cliniquement significatif ou important sur la qualité de vie liée à la santé.
Une série d'analyses en sous-groupes préspécifiés de la SSP a montré des résultats cohérents dans tous les sous-groupes de patientes par : âge (< 65 ans ou ≥ 65 ans), origine ethnique, région géographique, site de la maladie, résistance à l'hormonothérapie, présence d'une maladie mesurable, statut des récepteurs à la progestérone et statut ménopausique. Une réduction du risque de progression de la maladie ou de décès a été observée chez les patientes présentant une maladie viscérale (HR de 0,481 [IC à 95 % : 0,369 ; 0,627]), SSP médiane de 14,7 mois versus 6,5 mois) ; chez les patientes présentant des atteintes exclusivement osseuses (HR de 0,543 [IC à 95 % : 0,355 ; 0,833]) ; et chez les patientes avec une maladie mesurable (HR de 0,523 [IC à 95 % : 0,412 ; 0,644]). Chez les patientes en pré/périménopause, le hazard ratio était de 0,415 (IC à 95 % : 0,246 ; 0,698) ; chez les patientes négatives au récepteur à la progestérone, le HR était de 0,509 (IC à 95 % : 0,325 ; 0,797]).
Dans une sous-population de patientes présentant une maladie localement avancée ou métastatique qui n'ont pas été antérieurement traitées par une hormonothérapie, la SSP était également concordante.
L'analyse de la survie globale (SG) dans la population en ITT a montré une amélioration statistiquement significative chez les patientes traitées par Verzenios plus fulvestrant par rapport à celles ayant reçu le placebo plus fulvestrant. Les résultats de survie globale sont résumés dans le Tableau 12 et la Figure 5.
Tableau 12. Etude MONARCH 2 : Résumé des données de survie globale (Population en intention de traiter)
| | Verzenios plus fulvestrant | Placebo plus fulvestrant |
| Survie globale | N = 446 | N = 223 |
| Nombre d'événements (n, %) | 211 (47,3) | 127 (57,0) |
| SG médiane [mois] (IC 95 %) | 46,7 (39,2 ; 52,2) | 37,3 (34,4 ; 43,2) |
| Hazard ratio (IC 95 %) | 0,757 (0,606 ; 0,945) | |
| Valeur de p | 0,0137 | |
N = nombre de patientes ; IC = intervalle de confiance ; SG = survie globale
Figure 5. Etude MONARCH 2 : Courbe de Kaplan-Meier de la survie globale (Population en intention de traiter)
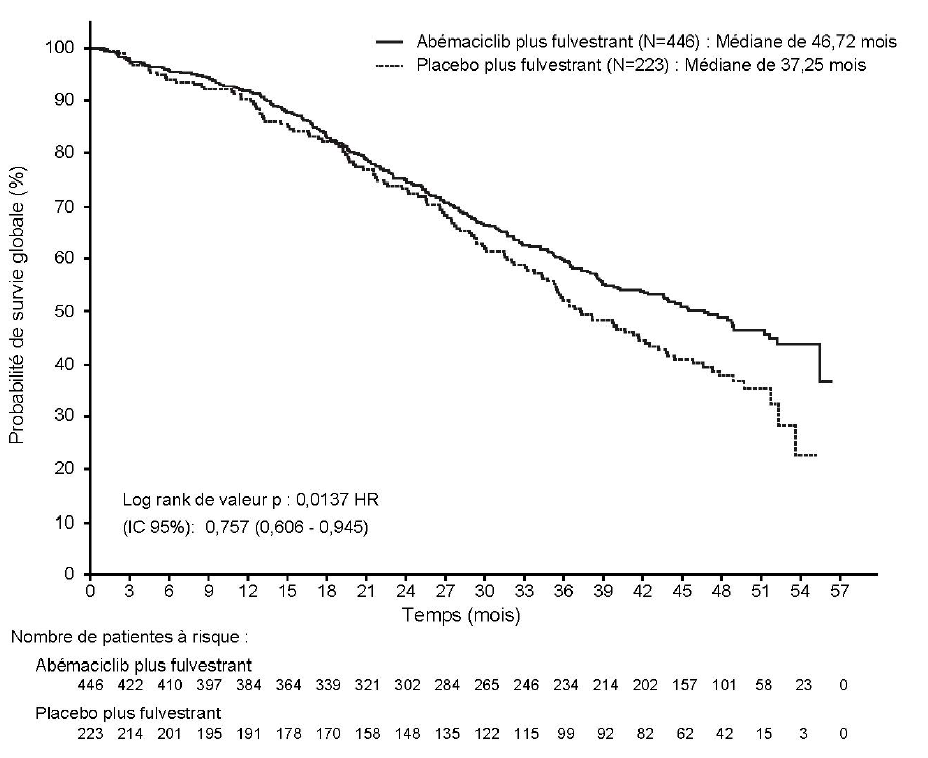
Les analyses de la SG par facteurs de stratification ont montré un HR de la SG de 0,675 (IC 95 % : 0,511 ; 0,891) chez les patientes avec une maladie viscérale, et de 0,686 (IC 95 % : 0,451 ; 1,043) chez les patientes ayant une hormonorésistance primaire.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Verzenios dans tous les sous-groupes de la population pédiatrique dans le cancer du sein (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
L'absorption de l'abémaciclib est lente, avec un Tmax de 8 heures et une biodisponibilité absolue moyenne d'environ 45 %. Dans l'intervalle posologique de 50 à 200 mg, l'augmentation de l'exposition plasmatique (AUC) et de la Cmax est approximativement proportionnelle à la dose. L'état d'équilibre a été atteint dans les 5 jours suivant une administration biquotidienne répétée, et l'abémaciclib s'accumule selon une moyenne géométrique du ratio d'accumulation de 3,7 (CV = 58 %) et 5,8 (CV = 65 %) en fonction de la Cmax et l'AUC respectivement. La consommation d'un repas riche en lipide a entraîné une augmentation de 9 % de l'AUC combinée de l'abémaciclib et de ses métabolites actifs et une augmentation de 26 % de la Cmax. Ces modifications n'ont pas été considérées comme cliniquement pertinentes. Par conséquent, l'abémaciclib peut être pris avec ou sans aliments.
Distribution
L'abémaciclib est fortement lié aux protéines plasmatiques humaines (fraction liée moyenne d'environ 96 % à 98 %). La moyenne géométrique du volume de distribution systémique est d'environ 750 L (CV = 69 %), ce qui indique la distribution de l'abémaciclib dans les tissus.
Les concentrations d'abémaciclib et de ses métabolites actifs dans le liquide céphalo-rachidien sont comparables aux concentrations plasmatiques libres.
Biotransformation
Le métabolisme hépatique est la principale voie d'élimination de l'abémaciclib. L'abémaciclib est métabolisé en plusieurs métabolites principalement par le cytochrome P450 (CYP) 3A4. La voie primaire de biotransformation est l'hydroxylation en un métabolite circulant avec une AUC correspondant à 77 % de l'AUC de la substance d'origine. De plus, les métabolites desethyl et desethylhydroxy circulent avec une AUC correspondant respectivement à 39 % et 15 % de l'AUC de la substance d'origine. Ces métabolites circulants sont actifs avec une puissance similaire à celle de l'abémaciclib.
Élimination
La moyenne géométrique de la clairance hépatique (CL) de l'abémaciclib était de 21,8 L/h (CV
= 39,8 %) et la demi-vie d'élimination plasmatique moyenne de
l'abémaciclib chez les patients était de 24,8 heures (CV = 52,1 %).
Après administration d'une dose orale unique de
[14C]-abémaciclib, environ 81 % de la dose a été excrétée dans les fèces et 3,4 % dans l'urine. La majorité de la dose éliminée dans les fèces était sous forme de métabolites.
Populations particulières
Age, sexe et masse corporelle
L'âge, le sexe et la masse corporelle n'ont eu aucun effet sur l'exposition à l'abémaciclib d'après une analyse pharmacocinétique de population menée chez des patients atteints de cancer (135 hommes et 859 femmes, âgés de 24 à 91 ans, et d'une masse corporelle comprise entre 36 et 175 kg).
Insuffisance hépatique
L'abémaciclib est métabolisé dans le foie. Une insuffisance hépatique légère (Child Pugh A) et modérée (Child Pugh B) n'a eu aucun effet sur l'exposition à l'abémaciclib. Chez les sujets présentant une insuffisance hépatique sévère (Child Pugh C), l'AUC0-8 de l'abémaciclib et la puissance ajustée de l'abémaciclib libre combinée à ses métabolites actifs augmentent respectivement de 2,1 fois et de 2,4 fois. La demi-vie de l'abémaciclib a augmentée de 24 à 55 heures (voir rubrique Posologie et mode d'administration).
Insuffisance rénale
La clairance rénale de l'abémaciclib et de ses métabolites est mineure. Une insuffisance rénale légère et modérée n'a eu aucun effet sur l'exposition à l'abémaciclib. Il n'y a pas de données chez les patients présentant une insuffisance rénale sévère, une néphropathie en phase terminale ou chez les patients sous dialyse.
Verzenios a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Il doit être conseillé aux patients d'être prudents lors de la conduite de véhicules ou l'utilisation de
machines s'ils ressentent une fatigue ou des sensations vertigineuses pendant leur traitement par Verzenios (voir rubrique Effets indésirables).
Les résultats sur les principaux organes cibles ayant une pertinence potentielle pour l'homme ont été observés dans le tractus gastro-intestinal, les organes hématolymphopoïétiques, et l'appareil reproducteur mâle chez les souris, les rats et les chiens dans des études d'une durée maximale de 13 semaines. Des effets sur les yeux et les valves cardiaques ont été observés uniquement chez les rongeurs à des niveaux d'exposition cliniquement pertinents. Des effets sur les poumons et le muscle squelettique n'ont été observés que chez les rongeurs à des niveaux d'exposition au moins 2 fois supérieurs aux niveaux d'exposition chez l'homme. Des effets sur le rein ne sont apparus que chez les rongeurs à des niveaux d'exposition au moins 6 fois supérieurs aux niveaux d'exposition chez l'homme. Une récupération complète ou partielle a été observée pour tous les organes cibles à la fin de la période de récupération de 28 jours, à l'exception des effets sur l'appareil reproducteur mâle.
Génotoxicité
L'abémaciclib n'a pas montré de potentiel mutagène au cours d'un test de mutation réverse sur des bactéries (test d'Ames), ni de potentiel clastogène au cours d'un test in vitro d'aberration chromosomique mené sur des lymphocytes du sang périphérique humain, et n'a pas non plus montré de potentiel clastogène au cours d'un test in vivo du micronoyau sur moelle osseuse de rat.
Carcinogénicité
La carcinogénicité de l'abémaciclib a été évaluée au cours d'études d'une durée de 2 ans menée chez des rats et des souris. Chez les rats mâles, l'administration quotidienne d'abémaciclib par voie orale a entraîné des adénomes testiculaires à cellules interstitielles bénins à des expositions correspondant à environ 1,5 fois l'exposition clinique chez l'homme. De plus, une hyperplasie des cellules interstitielles a été observée à des expositions correspondant à environ 0,1 fois l'exposition clinique chez l'homme. On ignore si ces effets seront transposables à l'homme. Aucune atteinte néoplasique liée à l'administration d'abémaciclib n'a été mise en évidence chez les souris ou les rates.
Détérioration de la fertilité
L'abémaciclib peut altérer la fertilité chez les mâles en âge de procréer. Dans des études de toxicité à doses répétées d'une durée allant jusqu'à 3 mois, les anomalies liées à l'administration d'abémaciclib observées au niveau des testicules, de l'épididyme, de la prostate et de la vésicule séminale ont inclus une diminution du poids des organes, la présence de débris cellulaires intratubulaires, une hypospermie, une dilatation tubulaire, une atrophie tubulaire et une dégénérescence/nécrose tubulaire. Ces effets ont été observés chez les rats et les chiens à des expositions correspondant à environ 2 et 0,02 fois l'exposition clinique chez l'homme, respectivement. Dans une étude de fertilité sur des rats mâles, l'abémaciclib n'a pas eu d'effets sur les performances de reproduction.
Dans une étude de fertilité et de développement embryonnaire précoce menée chez des rates, ainsi que dans des études de toxicité à doses répétées, l'abémaciclib n'a pas eu d'effet sur les performances de reproduction, ni d'effets importants sur l'appareil reproducteur femelle pouvant indiquer l'existence d'un risque d'altération de la fertilité chez les femelles.
Toxicité sur le développement
L'abémaciclib a été tératogène et a provoqué une diminution du poids du fœtus à des expositions des femelles correspondant à la dose recommandée chez l'homme.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament nécessitant une surveillance particulière pendant le traitement
Prescription hospitalière
Prescription réservée aux spécialistes et services en cancérologie et oncologie médicale
Comprimé ovale de 7,5 x 13,7 mm, jaune, avec l'inscription « Lilly » sur une face et le chiffre « 150 » sur l'autre face.
Plaquettes thermoformées en PCTFE/PE/PVC scellées avec une feuille d'aluminium dans un étui calendaire dans une boîte de 14 comprimés pelliculés.
Chaque comprimé pelliculé contient 150 mg d'abémaciclib.
Excipients à effet notoire
Chaque comprimé pelliculé contient 42 mg de lactose monohydraté. Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
croscarmellose sodique lactose monohydraté cellulose microcristalline silice colloïdale hydratée stéarylfumarate de sodium
Pelliculage
alcool polyvinylique (E1203) dioxyde de titane (E171) macrogol (E1521)
talc (E553b)
oxyde de fer jaune (E172)